


1Department of Dermatology and Allergy, Herlev and Gentofte Hospital, Hellerup, and 2Department of Dermatology, Aarhus University Hospital, Aarhus, Denmark
Atopic dermatitis is a prevalent inflammatory skin condition characterized by itch and dry skin, which affects 15–20% of children and 3–5% of adults. This article reviews epidemiological, clinical and experimental data to provide an overview of the most important disease mechanisms in atopic dermatitis. Genetic predisposition, environmental insults, atopic triggers, complex host immune response and skin barrier changes, and altered skin microbiota are discussed. Whilst our understanding of atopic dermatitis has improved dramatically in recent years, many basic aspects are still not understood. Further research is needed to fully understand this complex skin disease.
Key words: atopic dermatitis; aetiology; pathophysiology; pathomechanism; risk.
Accepted May 7, 2020; Epub ahead of print May 15, 2020
Acta Derm Venereol 2020; 100; adv00162.
Corr: Jacob P. Thyssen, Department of Dermatology and Allergy, Herlev and Gentofte Hospital, Hospitalsvej 15, DK-2900 Hellerup, Denmark. E-mail: jacob.p.thyssen@regionh.dk
The aetiology of atopic dermatitis is poorly understood, but studies have provided insight into the pathomechanism, which may improve the prediction of onset of atopic dermatitis and its prophylaxis. This review provides an overview of the pathogenesis and pathomechanism of atopic dermatitis.
Atopic dermatitis (AD) is a prevalent inflammatory skin condition characterized by itch and dry skin, which affects 15–20% of children and 3–5% of adults. In large proportions of affected patients AD is chronic or remitting, as shown by epidemiological studies (1).
The pathogenesis of AD is complex and poorly understood. However, in recent years, there has been major advancements in our understanding of the disease mechanism of AD, e.g. through the discovery of common filaggrin gene (FLG) mutations as a strong risk factor for AD, as well as the significant clinical effects of antagonistic therapy against interleukins (IL) 4, 13, 22 and 31.
This review provides a holistic overview of the most important disease mechanisms in AD.
AD predominately begins in early childhood, as indicated by a recent prospective Danish study, which showed that nearly all cases of AD are diagnosed before the age of 7 years (2). It is currently unclear to what degree “late-onset AD” is important in absolute numbers, as studies have shown that patients who present with AD in adulthood may have forgotten about their childhood AD, and that the disease may therefore represent re-activation of previous disease. This notion is strongly emphasized by the finding that approximately 29% of Swedish adults aged 31–42 years with a school health record of AD in childhood did not recall this when asked as adults (3). In asthma, patients with adult onset seem to have different disease mechanisms, and it is possible that this may also be the case for AD. Moreover, the epidemiology of AD may change over time, in concert with new causative exposures. As an example, use of cosmetic products in adolescence has been associated with new onset of AD or recurrence of previous disease (4). Nonetheless, AD normally begins in early childhood; a time where the skin barrier is vulnerable to stress (5–7). This will lead to a decrease in the threshold level against common triggers. As discussed in this review, the skin barrier defect is central to the risk of developing AD.
AD is a clinical syndrome, as indicated by the Hanifin & Rajka criteria for AD (8). These criteria dictate that a certain number of major and minor criteria need to be fulfilled in order to make a diagnosis of AD, including a list of phenotypic and heritable characteristics, such as xerosis, palmar hyperlinearity, keratosis pilaris (all associated with FLG mutations), infra-orbital folds or darkening, as well as facial pallor. Importantly, family predisposition to atopic disease is a major criterion of the Hanifin & Rajka criteria, and twin studies have shown that the heritability of AD is very high (9). The Hanifin & Rajka criteria were unintentionally developed for use in patients with predominately European ancestry, and it is clear that the phenotypic characteristics observed in other ethnic groups are under-represented, and that the criteria may fail when used in these populations (10). An example is the recent observation that pigmentation on the lips is associated with AD in Asian subjects (11).
FLG mutations lead to dry skin, characterized by elevated pH, increased colonization with staphylococci, enhanced penetration and reactivity to chemicals and allergens, and therefore, expectedly, a strongly increased risk of AD (12). Nearly all carriers of FLG mutations with AD develop their skin disease within their first 2 years of life (13), whereas children with later onset do not have these mutations (14). The discovery of FLG mutations provided a new, and much needed, basis for the study of paediatric AD, and led to a strong re-emphasis on primary skin barrier impairment as a crucial factor for the development of AD. Since then, it has been shown that dry skin at birth and at 2 months of age, independent of FLG mutations, can predict AD at 12 months of age, and that daily application of emollients in high-risk infants may reduce the risk of AD (15). Importantly, the normal skin barrier in the 2 first years of infancy is very different from that of adult skin; for example, the levels of natural moisturizing factors (NMF), a degradation product of filaggrin, are much reduced (16). The tendency for AD to begin on the cheeks is also explained by a local, very pronounced, reduction in NMF, which may last until 3 years of age (6). The down-regulation of filaggrin on exposed skin areas, as well as the increased prevalence of FLG mutations in populations that have migrated far from the Equator, is probably explained by evolutionary benefits due to increased synthesis of vitamin D following facilitated penetration of ultraviolet (UV) (17). Importantly, a deficiency of filaggrin, whether primary or secondary, results in increased penetration of allergens and risk of sensitization, which, in turn, may explain the increased risk of allergic asthma, rhinitis and food allergy in carriers of FLG mutations who have AD (18).
The crucial role of environmental exposure and skin stressors cannot be overemphasized when explaining the aetiology of the AD epidemic. Modern society has resulted in dramatic changes in human exposure, with increased use of, or exposure to, household products, cosmetics, tobacco, processed food, and air pollution, but at the same time reduced exposure to microorganisms and solar irradiation, as a result of increased hygiene, fewer people living together in the same household, and less time spent outside. Epigenetic changes due to environmental changes or insults could explain a large part of the endemic proportions of AD. In support of this theory, large genome-wide association studies have identified only a small proportion of genetic factors associated with AD (19). However, how the environmental changes have influenced the risk of AD at a mechanistic level is largely unknown.
Being born in the autumn or winter in the Northern hemisphere, or being exposed to a dry and cold climate, has been strongly associated with AD (20, 21). This is probably explained by skin exposure to low temperatures, as well as low ambient humidity due to indoor heating, which can negatively affect the skin barrier and result in dermatitis (22). Similarly, bathing infants in hard water may increase the risk of AD, possibly due to increased pH, which, among other aspects, results in premature cleavage of cornedesmosomes (20). Exposure to air pollution and being born in a newly built home have also been associated with AD (23, 24), perhaps because chemicals negatively affect the epidermal barrier. For example, short-term exposure to airborne formaldehyde results in increased water loss from the skin surface (25) in patients with AD, and toluene, a common air pollutant, can directly down-regulate synthesis of filaggrin (26). Interestingly, exposure to solar irradiation, which is normally avoided in infancy, to reduce the risk of skin malignancy, seems to protect against AD (27, 28). This could be explained by the positive effects of sub-erythemogenic doses of UVB irradiation on the skin barrier, which, among other aspects, reduces Staphyloccocus aureus colonization, itch, and T-cell invasion.
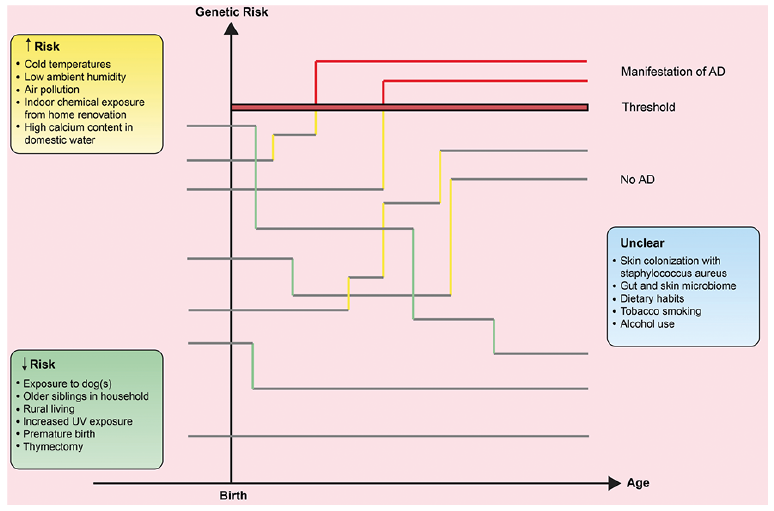
The crucial role of early-age alterations in immune activity on the development of AD is emphasized by the significantly reduced risk of AD in premature infants (29). Moreover, thymectomy in infancy reduces the risk of AD by 20%, suggesting that removal of the thymus decreases the number of circulating T cells that can act to develop AD (30). In indirect support of this assumption, a study found significantly larger thymus sizes in children with AD compared with controls, although this may also be a consequence of the increased demand for T cells in patients with AD (31). The farm theory suggests that microbial exposure may reduce the risk of diseases mediated by T-helper (Th) cell 2, including AD (32), but it is probably more important for allergic diseases than for AD per se. The finding that neonate exposure to dogs can strongly reduce the risk of AD could be confounded, but it is also possible that changes in the host gut microbiome can affect the tolerance-reactivity balance (33). It is unclear how nutrients and alcohol use in mothers can affect the risk of AD, but is has been suggested that the Th2 skew induced by alcohol intake may lead to a higher prevalence of AD in infants (34). Similarly, nutrients may affect the child’s immune response, but this area is complex, and little evidence exists. Collectively, AD occurs mainly in genetically predisposed individuals who have significant skin barrier impairment and who are exposed to AD triggers (or who are overly protected against the crucial microorganisms that could prevent excessive Th2 skew in childhood) (Fig. 1).
Fig. 1. Theoretical outline of how genetic risk genes and environmental risk exposures interact and may impact the risk of atopic dermatitis (AD). If a child reaches the threshold bar for AD, the disease will manifest. Factors that increase the risk of AD are represented by yellow vertical lines, whereas factors that decrease the risk are represented by green vertical lines. Once AD has manifested, the lines are shown in red.
AD is a skin condition in which primary (or secondary) skin barrier impairment leads to (further) skin inflammation, and in which S. aureus colonization may increase, and in turn may drive both eczema severity and the relentless sensation of itch (35). This leads to scratching and additional barrier impairment, thus creating a vicious cycle. Clinicians attempt to stop this cycle by restoring the skin barrier with emollients, reducing inflammation and itch with use of topical/oral immune suppressants or immune modulating drugs, as well light therapy, and, finally, decreasing the burden of S. aureus by use of disinfectants and antibiotics. Evidence supporting the benefits of emollient use to treat AD is the strongly increased time to subsequent flares in emollient users, and the reduced need for topical corticosteroids (36). However, barrier restoration without simultaneous control of inflammation seems to be inadequate in the treatment of AD (37). Prophylactic use of topical anti-inflammatory agents, e.g. with application twice weekly, works to reduce the risk of new flares (38).
While the exact role of bacteria in the pathogenesis of AD is unclear, colonization with S. aureus is very common in lesional and non-lesional AD skin. Antimicrobial peptides, which work as broad-spectrum antibiotics to kill Gram-negative and Gram-positive bacteria, are reduced in patients with AD, which, in turn, allows bacteria to colonize the skin (39). S. aureus can induce serine protease activity, which will destroy corneodesmosomes, and allow invasion (40). Moreover, the expression of Th2 cytokines is activated by proteases released by S. aureus (41), and S. aureus toxin increases the allergic response by activating mast cells (42), and induces up-regulation of T cells via a superantigen-mediated mechanism (43). S. aureus also release α-toxins, which forms pores in keratinocyte membranes leading to cellular damage (44). Individuals with AD and FLG mutations have a 7-fold higher risk of S. aureus skin infections, in part due to increased pH, but also due to the lack of the direct growth inhibition of the filaggrin proteins (45, 46). The levels of filaggrin degradation products, i.e. NMF, seem to regulate the strength of S. aureus corneocyte adhesion, the first step in skin colonization (47).
While the skin hosts the most diverse commensal community of humans, with over 1,000 different bacterial species, the role of the skin microbiome in AD is poorly understood (48, 49). An animal study showed that filaggrin deficiency and microbial dysbiosis triggered intracellular IL-1α secretion and drove chronic inflammation, hence indicating an important pathogenic role (50). Moreover, following successful treatment of AD, Streptococcus, Propionibacterium, and Corynebacterium species increase in numbers along with microbial diversity (51).
It is important to understand that non-lesional AD skin is also different from the skin of normal controls (Fig. 2). It shows decreased or altered synthesis of important epidermal proteins, e.g. filaggrin, filaggrin 2, involucrin, loricrin, hornerin, and tight junctions, but also decreased synthesis of antimicrobial peptides and lipids, (52–58) as well as increased expression of high-affinity IgE receptor on dendritic CD1a, along with increased numbers of T cells and their cytokines. Children with AD and food allergy have stratum corneum abnormalities in non-lesional skin that are not found in children with AD and controls without food allergy. Thus, filaggrin and ω-hydroxy fatty acid sphingosine are reduced, and there are important changes in the epidermal lamellar bilayer architecture (59). Thus, skin measurements in non-lesional AD skin show elevated pH, increased water loss from the skin surface, and increased penetration of chemicals (60). Moreover, AD skin displays a reduced reactivity threshold to exogenous stressors, such as skin irritants, allergens and S. aureus, in part due to the creation of resident T-cell populations (61–63). The changes in non-lesional skin are largely determined by disease extent and severity (53), probably reinforcing the impression of AD as a generalized skin disease.
Fig. 2. Important skin barrier changes in atopic dermatitis (AD). Innate and acquired inflammation in AD leads to downregulation and degradation of filaggrin and tight junction proteins, in turn leading to a dry and leaky skin barrier with elevated pH, which allows bacteria to colonize and allergens, irritants and microorganisms to invade. Tight junction reduction further allows antigen presenting cells to move upwards and meet the antigens. Lipid synthesis is compromised at several levels, which acts in concert with protein dysfunction to allow increased loss of water from the skin surface. In an attempt to restore the skin barrier and prevent excessive water loss, acanthosis occurs, often in conjunction with mild spongiosis.
Type 2 immunity-associated cytokines, such as IL-4 and IL-13, as well as other cytokines, including, but not limited to, IL-1, IL-17, IL-22, IL-31 IL-33, and thymic stromal lymphopoietin (TSLP) have important roles in AD. It is presently unclear whether significant differences exist between AD skin of children and adults, as well as between different ethnic groups, and to what degree this should affect treatment strategy (64, 65). While certain endotypes of AD are suspected to exist, the heterogeneous cytokine landscape could also, in part, be explained by the crucial pathogenic role of the sustained skin barrier impairment in lesional and non-lesional AD skin. Thus, the continuous bombardment and penetration of microorganisms, chemicals, irritants and allergens into the primary and sustained skin barrier impairment in AD could lead to secretion of various cytokines, and as discussed below, activate the Th1 and Th17 axis in addition to the Th2 axis. The exact immune response would be expected to depend on genetics, age, sites of skin exposure, possible co-infection, climatic effects, and type of elicitor. Interestingly, use of monoclonal antibodies against the IL-4 and IL-13 receptors seems to be slightly less effective in facial skin; an anatomical area which is exposed to environmental pollutants and climatic factors (66).
To date, there has been little research into the reactivity to various stressors. A survey in children with AD showed that sweating from exercise was a common exacerbator of AD (67). While the exact mechanisms is unknown (68) and, at least in part, could be explained by the direct effects of heating (69), leaking of sweat into the epidermis due to dysfunctional tight junction function could be relevant (70), as well as obstruction of sweat ducts due to filaggrin deficiency (71). Other well-established triggers for AD include exposure to wool, hot weather, psychological stress and sleep deprivation. Induction of stress leads to scratching behaviour in patients with AD, but not in controls (72). The dysfunctional and partly unresponsive peripheral hypothalamic-pituitary-adrenal axis in AD skin could also be important (73). Moreover, psychological stress reduces the recovery time of the stratum corneum, decreases lipid synthesis, and increases the risk of skin infections (74). Exposure to grass allergens may cause worsening of AD in grass-allergic AD individuals through IL-4 release (75). Contact allergens, e.g. fragrances and certain rubber chemicals, have been shown to elicit Th2 immune activity in patch test reactions, as opposed to many other allergens that elicit Th1 immune response (76, 77). Furthermore, exposure to experimental and environmental contact allergens in patients with AD causes Th2 immune response activity, but Th1 immune response in non-atopic skin (78). How this translates into clinical relevance is currently unclear. A recent study examined the skin immune response to various atopic triggers in individuals with normal skin and found that exposure to hard water is associated with IL-4 secretion in the epidermis (79).
It is beyond the scope of this review to describe the immunopathophysiology of AD in detail. Briefly, predominately Th2 (IL-4, IL-5, IL-13, IL-31) and Th22 (IL-22) deviation is observed in acute and chronic AD lesions, which, in turn, down-regulate expression of important skin barrier proteins, such as filaggrin. Innate lymphoid cells also release Th2 cytokines, now increasingly referred to as type 2 immunity. In chronic AD lesions, a parallel activation of the Th1 axis is observed, and in both acute and chronic AD, IL-17 activation can be found (82). Yet, even in healthy skin from patients with AD, there is increased expression of inflammatory cytokines and chemokines, as well as of their receptors, and an increased number of lymphocytes compared with healthy controls, suggesting increased immuno-surveillance in the skin and risk of acute inflammation (53).
Apart from the negative influence on the skin barrier, Th2 inflammation inhibits antimicrobial peptide synthesis and increases S. aureus colonization. The Th2 cells may, in many patients, lead to antibody isotype switching to IgE and recruit mast cells, eosinophils, basophils and dendritic cells. Elevated levels of IgE correlate with AD and atopic co-morbidities, including asthma and food allergies (83). Previously, this has been used to subtype AD into extrinsic AD, where allergic sensitization has taken place, and intrinsic AD, in which patients have normal levels of IgE. However, patients with normal IgE levels may also be sensitized and vice versa. It has even been suggested to use the terms intrinsic factors to describe inborn factors e.g. FLG mutations, Th2 skewing, etc., which affect the skin barrier function or the immune response in terms of AD and extrinsic factors to describe exogenous factors, e.g. S. aureus, detergents, allergens, etc. (82). Interestingly, IgE may target keratinocytes in up to 25% of patients with AD, indicating that IgE may play an important role in impairment of the skin barrier (84).
Regulatory T cells can suppress the Th2 response, and the balance between these 2 cell types is central to development of tolerance. It is not known whether a primary immune-deficiency/imbalance might be the prime cause of AD. Single nucleotide polymorphisms (SNPs) in, for example, ST2 (a member of the interleukin 1 family), IL-13, IL-12, have been reported to be associated with AD, and a huge work in developing a taxonomy for AD subtypes based on serum levels of cytokines has been undertaken (85). A recent work was able to distinguish at least 3 different subtypes of AD, based on analysis of 147 different soluble factors, yet this does not, in itself, show that the immune response is the prime cause of the disease (86). Rather, it indicates that patients with AD have different propensity to react to exogenous stimuli and that, even within the group of patients with AD, this differs slightly and gives rise to different subtypes. The result of this may be the development of personalized medicine for patients with AD (87).
Adult patients with AD have significantly elevated levels of circulating cytokines and chemokines (87). While it is intriguing to consider that the systemic inflammation in AD can negatively affect the function of other organs, such as the central nervous system and vascular system, there is currently no convincing evidence to support this. Nonetheless, AD has been associated with anxiety, depression, autism and attention deficit disorders, and it is possible that cytokines may cause a leaky blood–brain barrier and become absorbed into the cerebrospinal compartments and negatively affect cognitive development, by affecting the glia cells and neurogenesis. Decreased sleep quality due to itch is, however, also a major risk factor for ADD and depressive symptoms. The link between asthma and AD is not fully understood, but the shared type 2 immunity and effect of dupilumab on severity of both AD and asthma support that systemic inflammation could play an important role. While some patients with AD experience worsening of their AD during or after asthma attacks, it is unclear whether this is explained by psychological stress or by cytokines reaching the skin.
This review highlights some important disease mechanisms of AD. While understanding of AD has improved in recent years, many basic aspects are still not understood. For example, why do AD lesions outside the flexural areas tend to clear once flexural eczema is controlled? Why is AD a flexural disease? What triggers an AD flare? What explains the resolution of AD in the majority of children? What is the role of foods as triggers for AD? Why do AD children have fewer naevi than controls? These are just some of many unanswered questions. In conclusion, more research is needed into this complex skin disease.
Funding sources: JPT and MRR are financially supported by an unrestricted grant from the Lundbeck Foundation.
Conflicts of interest: JPT has attended advisory boards for Roche, Eli Lilly & Co., Pfizer, Abbvie, LEO Pharma, and Sanofi-Genzyme, been an investigator for Pfizer, Abbvie, Regeneron, Sanofi-Genzyme, and LEO Pharma, and received speaker honorarium from LEO Pharma, Abbvie, Regeneron, and Sanofi-Genzyme. CV has been investigator, speaker, or consultant for Novartis, Abbvie, Sanofi, LeoPharma and Eli Lilly & Co. MRR has no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize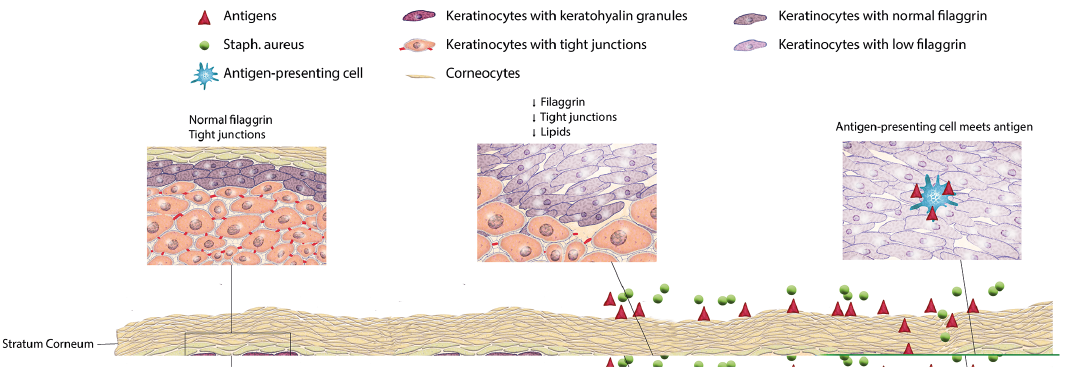 Click to show fullsize
Click to show fullsize